



Two-Photon Microscopy:
Principle and 3D Imaging Applications
Two-Photon Microscopy (2P microscopy) allows researchers to image deep inside biological samples. It is especially useful for studying thick tissues, 3D cell cultures such as spheroids and organoids, and even living organisms.
Unlike conventional fluorescence microscopy, two-photon microscopy uses near-infrared light to excite fluorescent molecules only within a very small, precisely defined focal volume. As a result, it produces high-contrast images with minimal background noise, enabling optical sectioning and deeper imaging of 3D samples.
What Is Two-Photon Microscopy?
Two-photon microscopy, also known as two-photon excitation fluorescence (TPEF) microscopy, is an advanced fluorescence imaging method based on the simultaneous absorption of two low-energy photons to excite a fluorophore. Unlike conventional microscopy, excitation occurs only in a highly confined focal volume. This reduces background fluorescence and eliminates the need for a confocal pinhole. The use of near-infrared light allows deeper penetration into biological tissues due to reduced scattering and absorption. As a result, two-photon microscopy is particularly suitable for imaging thick samples and living systems with high spatial resolution and minimal phototoxic effects.
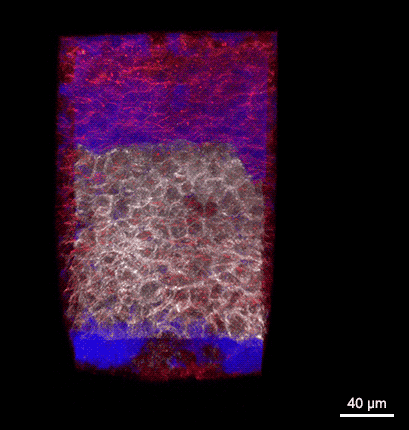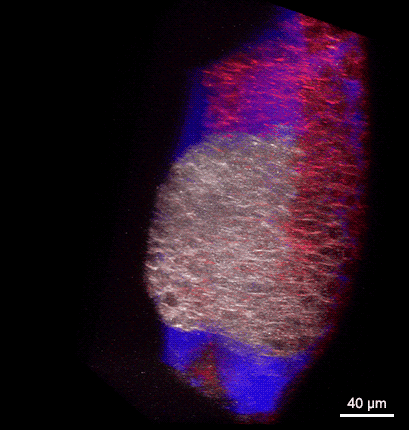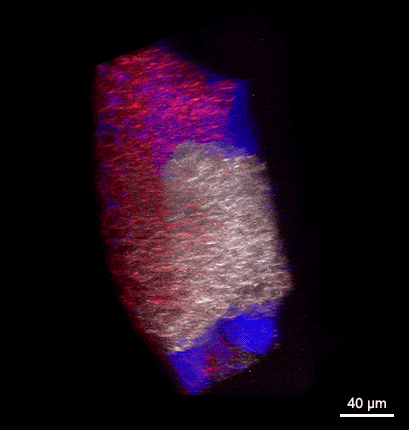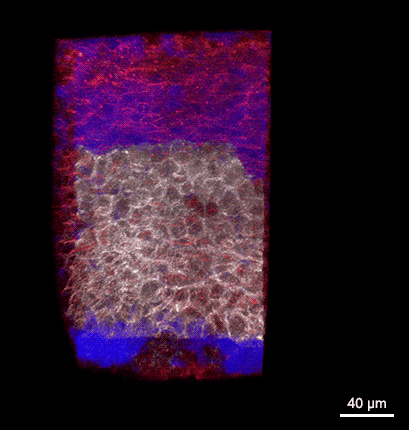
Rotating 3D rendering of a z-stack showing the dorsal germ ring of a zebrafish embryo at the onset of gastrulation (6 hours post fertilization). GFP (white) is expressed in internalizing prechordal plate progenitors and lyn-TagBFP (red) marks the membrane in all cells. The embryo was injected with dextran-rhodamine to label the interstitial fluid (blue). The image was recorded at the Bioimaging Facility of the Institute of Science and Technology Austria (IST), using a multiphoton LaVision BioTec TriM Scope microscope.
When to Use Two-Photon Microscopy
Use two-photon microscopy when:
- Imaging deep inside thick tissues (e.g., brain slices, tumors)
- Working with 3D cell cultures such as spheroids or organoids
- Performing live-cell or in vivo imaging experiments
- Reducing photobleaching outside the focal plane is critical
- Fluorescence imaging requires high contrast and low background
- Long-term time-lapse imaging under physiological conditions
For larger samples, alternative techniques such as light sheet microscopy are needed.
What Are the Main Applications of Two-Photon Microscopy?
Two-photon microscopy is widely used in life science research, particularly in situations where deep tissue imaging and minimal photodamage are required. It is especially well suited for in vivo imaging applications, such as observing neuronal activity in the brain of living organisms using cranial window preparations in mouse models, for example to track neuronal activity via calcium imaging assays (GCaMP). Additional applications include developmental biology, cancer research, and tissue engineering.
Why Is Two-Photon Microscopy Particularly Suited for 3D Imaging?
Two-photon microscopy enables deep imaging in biological tissues through a combination of optical advantages. It uses near-infrared light, which scatters less than shorter wavelengths and falls within the biological “optical window” (~650–1300 nm), where absorption is minimal. This allows light to penetrate deeper into samples.
In addition, excitation occurs only at the focal point, eliminating out-of-focus fluorescence. This significantly reduces background signals and photobleaching outside the imaging plane. Together, these properties allow high-resolution imaging at depths of several hundred micrometers, making two-photon microscopy especially well-suited for three-dimensional imaging of thick biological specimens.
Comparison: Two-Photon vs Confocal Microscopy
Two-photon and confocal microscopy differ in excitation method, imaging depth, and suitability for thick samples.
Use two-photon microscopy when:
- Imaging deep inside thick tissues (e.g., brain, tumors)
- Working with 3D models such as large spheroids or organoids
- Performing in vivo or long-term live-cell imaging
- Minimizing photodamage outside the focal plane is important
Use confocal microscopy when:
- Imaging 3D samples (up to ~100 µm) or cell monolayers
- Faster imaging is needed (depending on system, e.g., spinning disk)
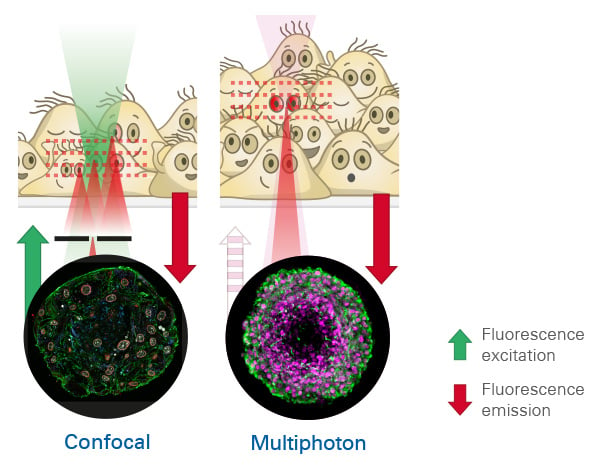
Comparison of confocal and two-photon (multiphoton) microscopy. Confocal microscopy (left) uses visible light, with limited penetration depth, and achieves optical sectioning by blocking out-of-focus light with a pinhole. Two-photon microscopy uses near-infrared light for two-photon excitation, enabling deeper tissue penetration. Restricted excitation to the focal point, results in intrinsic optical sectioning capability for 3D imaging.
| Feature | Two-Photon Microscopy | Confocal Microscopy |
| Excitation light | Near-infrared light (NIR) | Visible light (VIS) |
| Excitation region | Only at focal point | Along entire light path |
| Optical sectioning | Intrinsic (no pinhole needed) | Requires pinhole |
| Imaging depth | Deep tissue imaging (up to ~1 mm) | Limited depth (~100 µm) |
| Photobleaching | Reduced outside focal plane | Higher in thick samples |
How Does Two-Photon Microscopy Work?
Two-photon excitation is a nonlinear optical process where a fluorophore absorbs two photons nearly simultaneously. Each photon provides half the energy (double the wavelength) required for excitation. For example, a fluorophore such as GFP, which is excited at 470 nm using conventional fluorescence microscopy, can be excited at around 940 nm using two-photon excitation. This process uses near-infrared light (typically 700–1300 nm), allowing deeper tissue penetration compared to visible wavelengths.
Because excitation depends on the square of the photon density, it occurs only at the focal point where photon density is highest. As a result, two-photon microscopy provides intrinsic optical sectioning without the need for a pinhole (such as used in confocal microscopy), reducing out-of-focus signal and improving image contrast.
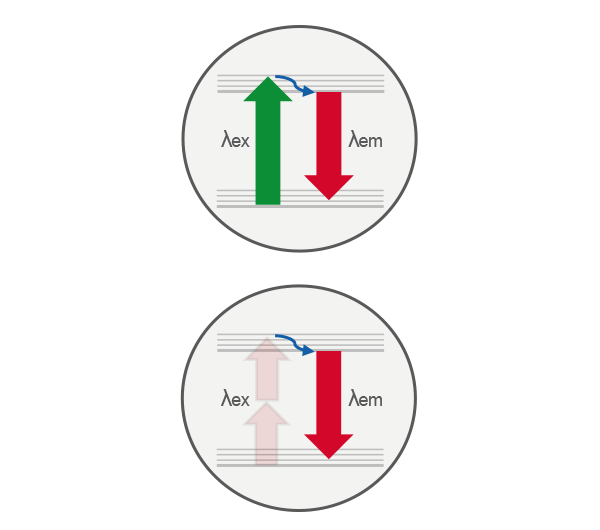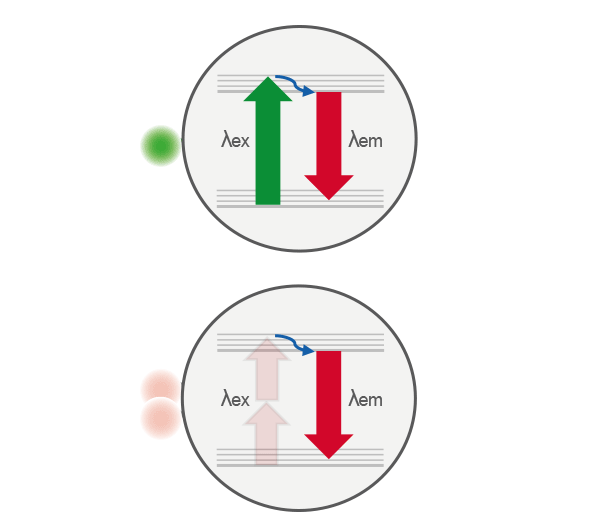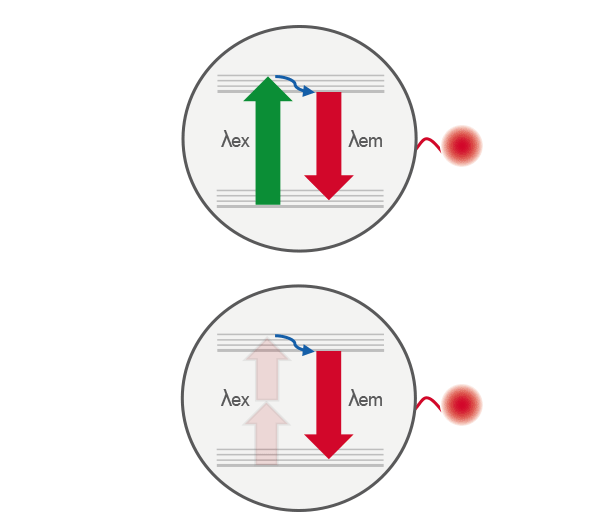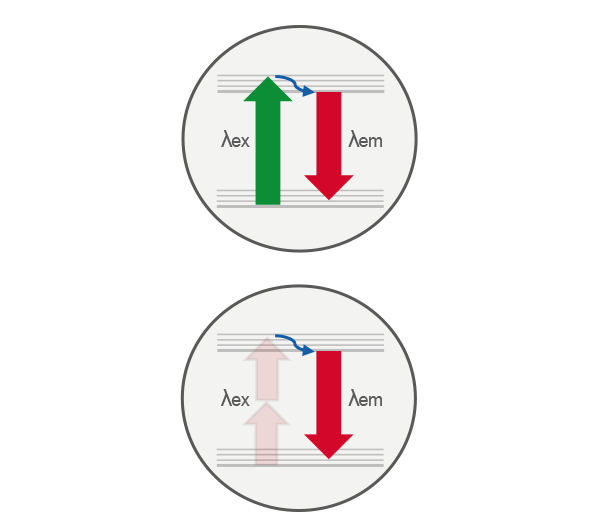
Comparison of single-photon (top) and two-photon (bottom) excitation. In single-photon excitation, a fluorophore absorbs one photon to reach an excited electronic state. In two-photon excitation, the same excited state is achieved by the near-simultaneous absorption of two lower-energy photons, enabling excitation only at the focal point where photon density is highest.
Advantages and Limitations of Two-Photon Microscopy
Two-photon microscopy enables deep tissue imaging using near-infrared light, which reduces scattering and allows imaging up to several hundred micrometers. Because excitation is confined to the focal volume, it provides intrinsic optical sectioning with high contrast and minimal background. In addition, photobleaching and photodamage outside the focal plane are significantly reduced, making the technique well suited for live-cell and in vivo imaging. It can also be easily combined with other nonlinear methods such as higher harmonics (SHG and THG).
However, two-photon microscopy requires complex and expensive laser systems. The excitation efficiency is lower, requiring high photon densities that may cause localized photodamage if not carefully controlled. Imaging speed is also limited due to point-scanning, which can be a disadvantage for fast processes.

Which ibidi Products Can Be Used for Multiphoton Microscopy?
All ibidi µ-Slides, µ-Dishes, and µ-Plates with an ibidi #1.5 Polymer Coverslip and the ibidi #1.5H Glass Coverslip are well suited for multiphoton microscopy, providing high optical quality and low autofluorescence for deep tissue imaging.
However, many multiphoton setups are build in an upright configuration and need labware that can be accessed from the top. In this context, ibidi µ-Dishes and µ-Plates or the µ-Slide 18 Well – Flat are often preferred, as they allow easy sample access and are compatible with water-immersion objectives, which are commonly used in multiphoton imaging.
What Is Special in the Optical Setup of a 2P Microscope?
Two-photon microscopy requires a specialized optical setup designed to achieve very high focal photon densities in a controlled manner.
Laser Light Source
The excitation light source is typically a femtosecond pulsed laser. Pulsed operation is essential because it allows the generation of extremely high peak power within very short time intervals. This balance is critical to enable efficient nonlinear excitation while minimizing photodamage and thermal effects in the sample. Typically, femtosecond (fs) pulsed lasers (∼100–150 fs pulse duration) with repetition rates on the order of 80–100 MHz are used.
The excitation wavelengths used in two-photon microscopy are typically in the near-infrared (NIR) range. Commonly used wavelengths include ~940 nm and ~1030 nm for fluorophores such as GFP and YFP, respectively. These wavelengths fall within the “optical window” of biological tissue (∼650–1300 nm), where absorption by water and endogenous chromophores is relatively low. For higher-order processes such as three- and four-photon excitation, even longer wavelengths can be used, typically up to ~1300 nm and again beyond ~1700 nm. The intermediate range (1300–1700 nm) is less suitable because strong water absorption leads to increased heating.
Highly Sensitive Detectors
Fluorescence signals are typically collected using highly sensitive detectors such as photomultiplier tubes (PMTs) or avalanche photodiodes (APDs). Depending on the setup, other options such as hybrid detectors (HyDs) may also be used. Because excitation is confined to the focal volume, there is no need for a confocal pinhole, and both ballistic and scattered emitted photons can be collected efficiently in either the forward or backward direction, depending on the sample properties.
What Other Non-Linear Techniques Can Be Used?
Three-Photon and Four-Photon Microscopy
Higher-order nonlinear excitation techniques, such as three-photon and four-photon microscopy, extend the capabilities of two-photon imaging even further. By using longer excitation wavelengths, scattering is reduced, enabling imaging at greater depths up to the millimeter range. Three-photon microscopy has been shown to enable high-resolution imaging of subcortical brain structures at depths exceeding 1 mm in vivo, particularly when combined with adaptive optics to correct sample-induced aberrations and maintain signal quality at depth. However, these methods are not yet widely adopted due to several practical limitations. The probability of higher-order excitation decreases significantly, requiring much higher peak powers and more advanced laser systems. This increases system complexity and cost, and can also raise the risk of localized photodamage at the focal point. In addition, suitable laser sources for these wavelength ranges are less commonly available.

Adaptive Optics for Deep Tissue Imaging
For imaging at greater depths, adaptive optics (AO) such as deformable mirrors or phase plates can be implemented to correct optical aberrations introduced by heterogeneous biological tissue. By compensating for these distortions, adaptive optics improves both signal intensity and spatial resolution in deep imaging applications, further enhancing the performance of two-photon microscopy in challenging samples.
Second and Third Harmonic Generation Microscopy
Multiphoton microscopy systems can be extended to detect additional nonlinear optical signals. Second harmonic generation (SHG) enables label-free imaging of highly ordered, non-centrosymmetric structures and is particularly well suited for visualizing collagen fibers in biological tissues. In SHG, two photons of the excitation wavelength (e.g., 1030 nm) are combined into a single photon with exactly half the wavelength (e.g., 515 nm), without energy loss. This contrasts with fluorescence, where emitted photons are red-shifted due to a Stokes shift, resulting in lower photon energy. SHG microscopy enables the visualization of structural components like collagen without the need for fluorescent labeling. Therefore, applications comprise label-free monitoring of changes in the e.g., the collagen microenvironment in pathological conditions, such as tumor-associated matrix remodeling.
Third harmonic generation (THG) provides contrast based on refractive index changes and interfaces within the sample, allowing label-free imaging of structural features such as lipid boundaries. These complementary imaging modalities significantly expand the range of applications beyond fluorescence-based techniques.
Common Problems and Troubleshooting
Why is my fluorescence signal so weak?
A weak signal is often caused by low fluorophore concentration, poor staining, or inefficient excitation.
Solution: Increase laser power carefully, optimize your staining protocol, and verify fluorophore compatibility with two-photon excitation wavelengths.
Why do I see photodamage at the focal point?
High peak intensities required for two-photon excitation can lead to localized photodamage or bleaching at the focal spot.
Solution: Reduce laser power, shorten dwell time, and optimize scanning speed. Use more photostable fluorophores and avoid unnecessary repeated scanning.
Why do my fluorescence channels overlap?
Spectral overlap occurs when the emission spectra of different fluorophores overlap, causing signal from one fluorophore to be detected in multiple channels.
Solution: Choose fluorophores with well-separated emission spectra, adjust detection windows, use sequential scanning, or apply spectral unmixing to accurately separate the signals.
Why do I see stripes or shifted lines in my images?
Stripe patterns or line offsets are often caused by misaligned or improperly calibrated galvo scanners.
Solution: Check scanner calibration and alignment, verify scan settings, and perform system calibration routines. If the issue persists, contact technical support for optical realignment.
Why is my signal suddenly weak even though I didn’t change my sample preparation?
A sudden signal drop is often caused by laser power loss, dirty optics, or system misalignment.
Solution: If you suspect a laser or alignment issue, contact a qualified technician.
Attention! Two-photon microscopy uses light invisible to the human eye. High-power pulsed lasers can cause irreversible eye damage. Do not open the system or attempt internal adjustments!
FAQs
What is the difference between two-photon and widefield fluorescence microscopy?
In widefield fluorescence, the fluorescence intensity scales linearly with excitation intensity. The entire sample is illuminated, generating fluorescence both in and out of focus, which leads to background signals and reduced contrast. In contrast, two-photon microscopy is a nonlinear process, in which fluorescence scales quadratically with excitation intensity, restricting excitation to the focal volume and thereby strongly reducing out-of-focus fluorescence. Because of the higher tissue penetration and intrinsic optical sectioning, two-photon microscopy is well suited for 3D imaging, whereas widefield microscopy is typically limited to adherent cells or thin tissue sections.
What is the difference between two-photon and confocal fluorescence microscopy?
Confocal microscopy uses a pinhole to reject out-of-focus light, whereas two-photon microscopy relies on an intrinsically confined focal volume due to its nonlinear excitation. Two-photon excitation typically uses near-infrared light, which penetrates deeper into tissue, and requires very high peak intensities, so pulsed femtosecond lasers are used instead of continuous-wave lasers. This combination restricts fluorescence to the focal point, reduces photodamage outside the focal plane, and makes two-photon imaging especially suitable for in vivo imaging or thick biological samples.
What is the penetration depth using 2P microscopy?
Two-photon microscopy can reach depths of about 500–1000 µm in biological tissue, depending on the sample and fluorophore labeling, because it uses longer near-infrared excitation wavelengths that scatter less and penetrate deeper than visible light.
What can I do to reach an even higher penetration depth?
To achieve an even greater imaging depth, adaptive optics (AO) such as deformable mirrors or phase plates can be used to correct sample-induced aberrations and improve signal quality at depth. Additionally, tissue clearing techniques reduce light scattering by homogenizing refractive indices, enabling significantly deeper optical penetration.
Which fluorophores can be used for 2P microscopy?
Most standard fluorophores can be used if they have suitable two-photon absorption cross-sections. Common examples include GFP-like proteins, fluorescein derivatives, and rhodamine dyes.
References
Göppert-Mayer M. Elementary processes with two quantum transitions. Ann Phys. 1931; 9:273–294. doi: 10.1002/andp.19314010303
Denk W, Strickler JH, Webb WW. Two-photon laser scanning fluorescence microscopy. Science. 1990; 248:73–76. doi: 10.1126/science.2321027
Helmchen F, Denk W. Deep tissue two-photon microscopy. Nat Methods. 2005; 2:932–940. doi: 10.1038/nmeth818
Xu C. Multiphoton fluorescence microscopy for in vivo imaging. Cell. 2024; 187. doi: 10.1016/j.cell.2024.08.030
van Huizen LMG, Kuzmin NV, Barbé E, et al. Second and third harmonic generation microscopy visualizes key structural components in fresh unprocessed healthy human breast tissue. J Biophotonics. 2019; 12:e201800297. doi: 10.1002/jbio.201800297
Sohmen M, et al. Optofluidic adaptive optics in multiphoton microscopy. Biomed Opt Express. 2023; 14:1562. doi: 10.1364/BOE.480000
Streich L, et al. High-resolution structural and functional deep brain imaging using adaptive optics three-photon microscopy. Nat Methods. 2021; 18:1253–1258. doi: 10.1038/s41592-021-01257-6
Luu P, Kner P. More than double the fun with two-photon excitation microscopy. Commun Biol. 2024; 7. doi: 10.1038/s42003-024-06057-0

Article written by Stefanie Kiderlen, PhD
ibidi GmbH | April 30, 2026
Cell biologist and microscopy specialist with expertise in advanced imaging, live cell imaging, and 3D cell culture models. Stefanie received her PhD at the Biophysical Department at LMU, Munich, focusing on atomic force microscopy in 2D and 3D cell culture systems.